
Status quo
They contain 37 genes, they are found inside our cells, they are reproduced by binary division, possibly before symbiosis with ancient eukaryotes they lived as independent organisms, and in reproductive biology it is already possible to use them from a healthy donor. Yes, they are mitochondria.
By the final oxidizing the products of metabolism of carbohydrates, fats and proteins, mitochondria supply cells with energy in the form of ATP molecules. Thanks to mitochondria homo sapiens are able to think, to move in space, or, in your case, to read this post. And pathological mutations in these 37 genes (as well as in a nucleus of cells which are associated with mitochondria) lead to a disruption of the energy processes of the cells, this manifests as mitochondrial diseases.
For example, replacement of just one molecule, the base of adenine with guanine at position number 3243 in mitochondrial DNA, causes the syndrome called MELAS, which is “mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes”. Researchers from Peking University Chang-Yu Xia et al. studied cases of 100 pediatric patients with this mutation and found that the patients had one or more of the following symptoms: seizures, muscle weakness, headache combined with vomiting, blurred vision, hearing loss, impaired verbal communication, cardiac rhythm pathologies [1].
Nonspecific symptoms in mitochondrial diseases are explained in next: with such mutations, there is a dysfunction of cell metabolism, therefore, the most volatile tissues and organs “ suffer” first. They are nervous and muscular tissue, including the heart, the organs of hearing and vision, the gastrointestinal tract [2].
In a medical environment, mitochondrial diseases are considered to be an “any symptom, any age, any organ system, and any mode of inheritance” which does not make it easier for doctors to diagnose such diseases. In March 2018, an article was published by researchers from the Columbia University Medical Center which describes what such patients go through before receiving the final diagnosis [3]. I’ll write briefly about it.
How they were investigated?
Researchers contacted patients from the Rare Diseases Clinical Research Network, proposing an online survey of 25 non-mandatory questions to patients or caregivers. The average expected time for filling out the questionnaire is 15 minutes. Wherever possible, “yes/no” selection or lists were used, but an “other” answer option with text input was also included. The informed consent was obtained from patients, passport data was not recorded in the survey. 215 people filled the survey.
What diagnoses were noted by patients?
More often (21.8%) it was myopathy, 9.6% of cases was chronic progressive external ophthalmoplegia, 9.1% — MELAS, 8.1% — Leigh disease. One patient noted encephalopathy (0.5%), the “other” option was chosen by 28.4% of patients and usually they wrote biochemical deficiencies, more rare syndromes or secondary mitochondrial diseases, where mutations related to mitochondria is an important, but not the only reason in the development of the disease.
Which symptoms were noted?
Weakness (62.4%), fatigue (56.2%), difficulty walking (38.6%), droopy eyelids (32.9%) and impaired coordination (32.4%). Nonspecific complaints explain the difficulties in diagnosing.
Who was the first doctor to discuss these symptoms?
More than half of the patients first discussed the symptoms on the consultation with a general practitioner, 35.2% with a specialist. However, only 12.9% of the patients were provided with mitochondrial disease diagnosis from the first doctor. On average, such patients visited ~8 doctors before diagnosis (median value is 5). 7 patients, or 3.6%, visited 20 doctors or more.
Which specialists provided the diagnosis?
In 55.2% of cases it was a neurologist, in 18.2% — a clinical geneticist, in 11.8% — a specialist in metabolic diseases. This data correlates with the symptoms described above.
What diagnostic procedures were performed?
84.8% of the patients were investigated by blood tests (lactate, pyruvate, amino acid concentration, or genetic testing), 71% of patients had muscle biopsy with histological examination, 60.5% of patients had MRI, 38.6% tested for urine organic acids.
Which genetic tests?
39.5% of patients underwent sequencing of mitochondrial DNA prior to diagnosis, in 19% cases it was a sequencing of nuclear DNA, and only 11.4% received whole-exome sequencing.
Further actions
Most of the patients: — noted that they began a treatment after receiving the diagnosis (81.7%); — joined a patient organization or a community with mitochondrial diseases (71.8%). Almost all of them reported that participation in such organizations was beneficial for them;
On the way to diagnosis
More than half of the patients (54.6%) indicated that before the diagnosis of the mitochondrial disease, an alternative diagnosis was obtained, and 31.9% reported for having received more than one diagnosis. More often psychiatric syndromes (depression and conversion disorder), fibromyalgia, chronic fatigue syndrome, multiple sclerosis diagnoses were provided instead of the actual mitochondrial disorders.
“If you were to learn today that the mitochondrial disease diagnosis you received is incorrect, and that you do not in fact have a mitochondrial disease, how would this affect you?“
This question was asked to patients, and ~35% of them chose the option “positively” or “very positively”, ~30% noted that it would not have affected, and the for rest it would affect negatively. The authors believe that a significant number of patients who chose “neutrally” considered this answer because of a lack of a specific treatment for the diagnosed pathology.
As the result
Some weaknesses were present in the study, the main one: the information about diagnoses and symptoms was entered by patients themselves and this may compromise the results. Some patients do not take therapy, probably because they have not been offered therapy for their disease yet. And even among those who have received treatment prescriptions, just a symptomatic therapy may be implied. Patients consider it important to unite in organizations, since this implies the exchange of information with other patients and doctors, also their interests could be protected better. The authors note that in recent years a muscle biopsy, a painful and invasive method, is used less often, but the portion of genetic tests, such as sequencing of the mitochondrial genome, is increasing. The study makes it clear what patients with mitochondrial diseases go through, including the exceptional cases when the patient had to visit more than 20 doctors before the final diagnosis will be made. Probably, the situation with this group of diseases will improve with a reduction of the cost of genetic testing, in particular, the sequencing of the mitochondrial genome. There are great hopes that timely diagnostics will be achieved through the implementation of projects on creation of genetic passports at national levels. The therapy of such diseases, which is connected with the high cost of development and limited sales market, remains a problematic issue. This, in a long run, increases the financial burden on patients and other taxpayers in countries where orphan disease therapy is compensated for by public funds. Fortunately, in the post-genomic era, such patients have a chance to control their disease.
How do you think, are there people with such mutations that have not yet been identified among the people you know? And if you are a doctor — how do you think, how many of the mitochondrial diseases you did not recognize during first consultations?
References:
- Chang-Yu Xia et al., Clinical and Molecular Characteristics in 100 Chinese Pediatric Patients with m.3243A>G Mutation in Mitochondrial DNA. https://www.ncbi.nlm.nih.gov/pubmed/27503020
- Amy Goldstein Mitochondrial Diseases http://www.childneurologyfoundation.org/disorders/mitochondrial-diseases/
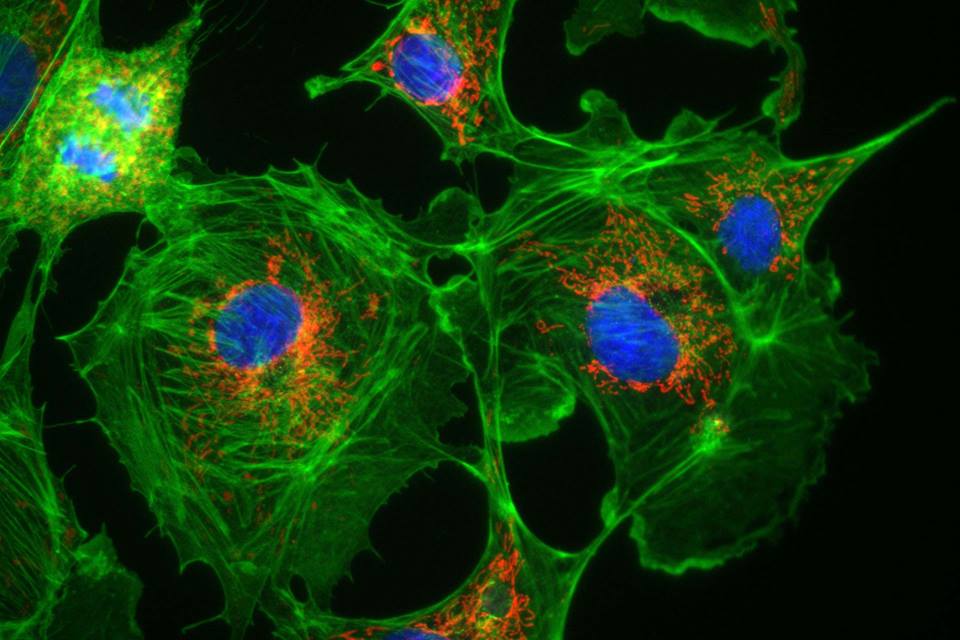
- Grier J et al. Diagnostic odyssey of patients with mitochondrial disease: Results of a survey. https://www.ncbi.nlm.nih.gov/pubmed/29600276 Image: Cells (BPAE) showing Mitochondria (red), actin filaments (green) and nuclei (blue) captured with iXon 897.Courtesy of Mike Davidson, Florida State University, USA; http://www.andor.com/cameras-for-microscopy
Additional information on the topic:
- Mitochondrial disease news https://mitochondrialdiseasenews.com/
- Foundation for Mitochondrial Medicine http://mitochondrialdiseases.org
- Rare Mitochondrial Disorders Service (UK) http://mitochondrialdisease.nhs.uk/
- Saskia B. Wortmann et al. Whole exome sequencing of suspected mitochondrial patients in clinical practice https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4432107/
- Vento JM Genetic counseling in mitochondrial disease. https://www.ncbi.nlm.nih.gov/pubmed/23264099
- Elizabeth McCormick et al. Molecular Genetic Testing for Mitochondrial Disease: From One Generation to the Next https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3625386/
- Carroll CJ Next-generation sequencing for mitochondrial disorders. https://www.ncbi.nlm.nih.gov/pubmed/24138576